EU product Registration Road Map
by Nicole Ann Piscopo Quality Assurance, Pharmacovigilance Specialist and GxP Auditor @PQE Group
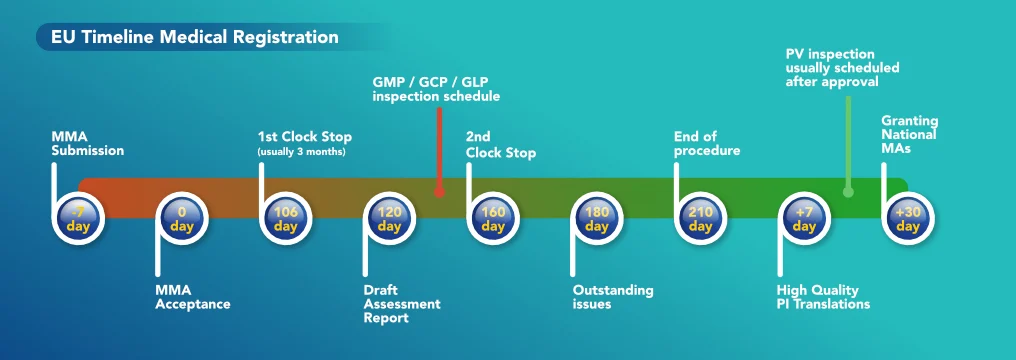
Case Study
With the increase in the demand for EU certified products, companies coming from non-regulated markets are searching for help in order to obtain this certification for their product. These inter-related services include maintenance of Good Manufacturing Practices and Good Distribution Practices, Quality Assurance, Qualified Person and Responsible Person services and Pharmacovigilance.
In order for a company which is located in a non-EU country to sell its products to another company in the EU, the product has to be EU certified. There are a series of steps which the manufacturing company must do in order to obtain the EU certification for its product. The first step the company needs to take is to Register the Dossier within the EU for the specific drug it intends to sell. The dossier is a complex document which contains all the details of the drug. Such details include the name of the drug, excipients, API, manufacturing processes, testing requirements, Qualified Person details, where the drug is registered, Pharmacovigilance details, MA number, etc. Two very important sections within this dossier are the manufacturing site (that must have the EU-GMP certification) and the EU batch testing and release site.
Interoperability: next steps
Once the dossier is submitted it automatically triggers an EU Authority Inspection. This allows the manufacturing company to seek professional consulting assistance in order to become EU-GMP compliant, which will work along with the manufacturing company to achieve its goal, which is to sell an EU-Compliant Product. An audit can be performed which will enable the manufacturing company to assess its inspection readiness. This audit can be used as an internal audit to review all the documentation, processes and implementation of Good Manufacturing Principles in order to identify gaps in the system. Consultancy services can also aid the manufacturing company in building and/or updating their Quality Management System. Once this is done, the system is considered EU-GMP compliant and the Authorities will be satisfied with the progress of the company and issue the EU-GMP certificate. When this certificate is issued the manufacturing site is at the same level as an EU manufacturing site.
Implementing the ISO IDMP Standards
Since the site is now EU-GMP certified, the product can be manufactured, tested and released from the manufacturing site. Although the product was released from a non-EU manufacturing site it still needs to be released within the EU, therefore, a representative sample is sent to a laboratory which is located in the EU in order to be retested and released. Once testing is concluded from the laboratory, the results are satisfactory and they comply with the results obtained from the non-EU manufacturing site, the batch can be released along with the Certificate of Analysis by the testing laboratory. The Certificate of Analysis contains all the specifications for the product along with the results obtained.
With the Certificate of Analysis in hand, a Batch Release Certificate is issued. This Batch Release Certificate must be issued prior to the product entering the supply chain and is used to apply to the respective Health Authorities for the Certificate of Pharmaceutical Product. This certificate is required by the importing country in order to register and distribute the product within that specific country. Once all this is completed, the manufacturing company can sell the product to its clients as an EU-certified product.
European Union Qualified Person for Pharmacovigilance (EU-QPPV)
The European Union states that each drug which is registered within the EU must have a European Union Qualified Person for Pharmacovigilance (EU-QPPV). Pharmacovigilance is used to monitor the drug and report any adverse effects the patient might experience when it is on the market. The EU-QPPV details also must be included in the initial submission of the dossier; this is required by the EU since the product will be registered within the EU. There is a set of qualifications one must have in order to be listed as an EU-QPPV, such as the EU-QPPV must reside in an EU country, Norway, Iceland or Liechtenstein and The EU-QPPV must be in possession of a university degree in pharmacy, medicine, veterinary medicine, chemistry, pharmaceutical chemistry and technology or biology.
The EU-QPPV needs to be contactable by any of the European Authorities for the drugs for which he/she is responsible. Even if the drug is not on the EU market, the EU-QPPV needs to be listed in the initial submission of the dossier and the drug must be monitored. EudraVigilance is a platform used to register drugs and to report/evaluate any suspected adverse reactions from the drugs taken by patients. The drug needs to be registered on the EudraVigilance and then linked to the EU-QPPV.
A pharmacovigilance quality management system is required and must be continuously kept up to date. A Pharmacovigilance Master File (PSMF) is to be created to explain the pharmacovigilance system which is used by the Marketing Authorization Holder. This must be located where the EU QPPV works or where the main activities of pharmacovigilance are carried out.
It is the responsibility of the EU QPPV to ensure that all adverse drug reactions (e.g., a patient ingested the drug and experienced a negative symptom), product quality complaints (e.g.,the patient did not inject the drug however there is a problem with it, such as the label on the bottle is torn) or medical inquiries (these are any questions about the drug which are not ADRs or PQCs, e.g., can I drink orange juice with the drug?) are logged.
Quintian Pharma can support all of these inter-related services and requirements, for both regulated and non-regulated markets. We are committed to providing services and support to the pharmaceutical industry, following all EU directives on new products.