Method Development in the EU
Introduction to Method Development
As a hard and fast rule, all analytical procedures in a pharmaceutical testing environment must follow a clear, easy to follow procedure that has been shown to be effective and applicable to the analyte in question, able to reliably and consistently produce the desired results.
Method development is a key process in the pharmaceutical industry that encompasses the process of establishing chemical analytical techniques to:
- Identify and/or quantify impurities, target molecules, residual solvents and degradants;
- Ensure critical quality parameters through quality control;
- Assess purity and potency of manufactured products.
Analytical methods are usually applied to:
- Raw material testing;
- In-process controls;
- Intermediate and final product testing;
- Cleaning processes;
- Stability testing.
The development of analytical procedures usually follows the life cycle below:
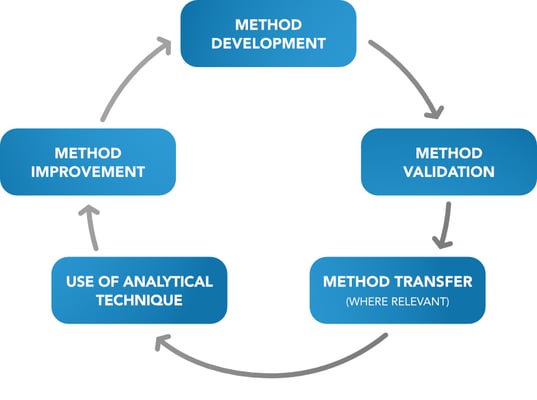
Method development parameters
Commonly used chemical analytical techniques in the pharmaceutical industry are generally chromatographic (HPLC, LC/MS, GC/MS, etc.), spectroscopic (UV, IR, MS, etc.) or fall under a third, less easily characterized category (TOC, PSD, etc.). As any analytical chemist will tell you, these techniques are never applied with a ‘one-size-fits-all’ approach.
The ways in which these techniques are applied will be highly dependent on a multitude of factors:
- The analyte itself: different analytes will have different physicochemical properties, which will in turn greatly affect the method development process. Using HPLC as an example, which is likely the most widespread analytical technique in quality control labs, intrinsic factors such as solubility, pKa, and chemical/thermal stability will influence choice and composition of mobile and stationary phases, flow rate and column temperature.
- Sample preparation: sample preparation is an essential part of chromatographic and spectroscopic analyses. The process is intended to provide a representative, reproducible, and homogenous solution that will provide analyses that are free from interference, compatible with the selected equipment, produce minimal peak shape distortions, and are free from background effects.
- Environmental conditions and equipment used: while a validated analytical method may be applicable to the laboratory that developed it, it may not be applicable to another laboratory, even when the method used is exactly the same. Different labs use different analytical instruments that are in different stages of qualification and maintenance, have different environmental factors such as ambient temperatures (even though these are controlled within pharmaceutical laboratories), and are used by different analysts to those who initially developed the method.
Method validation and transfer
Once methods are developed, they must be validated to produce documented evidence that results are accurate and reproducible. As with other validation activities, validation execution must be carried out according to a pre-defined protocol, derived from a validation master plan.
Within the EU, analytical method validation is often carried out in accordance to ICH harmonized guidelines: validation of analytical procedures Q2(R2). This serves as a general guideline to most validation activities (not as applicable to biotech or organic products) and are mostly focused on chromatographic techniques. Nevertheless, it lays out the minimum parameters which validation activities must reach:
- Accuracy
- Precision (including repeatability and reproducibility)
- Specificity
- Linearity
- Range
- Robustness
- Limit of quantification and detection
- Robustness (depending on analytical technique)
Method development in the EU
Most analytical methods applied to products released in the EU are laid out in the European Pharmacopeia (Ph. Eur.) which is a compendia of common, compulsory quality standards for all medicinal products in Europe. The Ph. Eur. is composed of 4 main chapters:
- General notices – instructions to understand texts, conventional expressions used throughout the text;
- General chapters – containing methods of analysis, materials for containers and containers and reagents;
- General monographs;
- Individual monographs
QC labs performing method development for products which already have a marketing authorization in the EU can follow the methods published within the Ph. Eur., however it still must be shown that the methods are applicable via a method verification. Method verification for compendial methods documents that the said method applies to that particular lab. If verification fails, then it is necessary to develop and fully validate a new method that deviates from the compendial method, and can be proposed for publication in the PH. Eur. Method verification for compendial methods are not necessary for all quality control tests – less complex tests do not require verification.
For preparations without an existing marketing authorization in the EU, methods must be developed and validated ‘from scratch.’ Testing limits must be based upon efficacy, toxicology considerations and analytical method capabilities, and must be supported by batch data.